
- Sustainability
- DE&I
- Pandemic
- Finance
- Legal
- Technology
- Regulatory
- Global
- Pricing
- Strategy
- R&D/Clinical Trials
- Opinion
- Executive Roundtable
- Sales & Marketing
- Executive Profiles
- Leadership
- Market Access
- Patient Engagement
- Supply Chain
- Industry Trends
Can Synthetic Control Arms Address Regulatory and HTA Evidence Requirements?
Real-world evidence (RWE) provides a unique opportunity to understand patient care and outcomes with more clarity than what is possible with traditional randomized clinical trials. As clinical trial design is evolving, synthetic control arms1 are playing an increasingly important role in product evaluation, but how do regulators and payers view these model-based control arms?
To explore this issue, Pharmaceutical Executive, in collaboration with Parexel, held a virtual roundtable to stimulate conversation about the opportunities that synthetic control arms offer for delivering real benefit to patients and product developers and how to overcome potential barriers with the use of this burgeoning clinical trial model. Participants in the roundtable were:
Lisa Henderson (Facilitator), Editorial Director, Pharmaceutical Executive
Leanne Larson (Moderator), Senior Vice President and Head of Real-World Evidence and Access, Parexel
Nuwan Kurukulasuriya, Senior Vice President and Head of Global Medical Affairs, MorphoSys
Amy McKee, Vice President and Head of Regulatory Consulting and Oncology, Parexel
Sangeeta Budhia, Global HTA Strategy Lead and Vice President of Pricing and Market Access, Parexel
EFFECTS OF COVID-19 ON CLINICAL TRIAL DESIGN
Leanne Larson, Senior Vice President and Head of Real-World Evidence and Access at Parexel, kicked off the discussion by reflecting on comments made by Amy Abernethy, MD, PhD, Principal Deputy Commissioner and Acting CIO at the FDA. In May 2020, when groups were starting to collaborate on how best to address the growing COVID-19 pandemic, Abernathy said that we need to learn from every patient around us and from all the data around us, and we need to learn quickly what works and what doesn’t.
“I think these comments are really important and drive home the fact that we’re currently witnessing and participating in an important evolution in clinical development,” said Leanne.
By that, Leanne explained that the pandemic made it clear that a drug development model that best incorporates all relevant available data will help sponsors make informed decisions and bring new therapies to patients in need more quickly. Building this model requires us to rethink long-held perspectives on clinical development, however.
“It’s time to think beyond the two well-controlled, randomized clinical trials (RCTs) model that has been at the core of drug development for so long, and to consider how RWE might answer other key questions in clinical research,” Leanne stated, adding that such information will help us better understand patient experiences and outcomes—and help us design and conduct better clinical trials.
Importantly, this kind of a learning system necessitates a critical feedback loop, with one piece (RCT) informing the other (RWE) and vice versa (Figure 1). For instance, RCT data must be tested in the real world in larger, more heterogeneous populations to ensure that we truly understand what will happen when patients are treated in routine care. Meanwhile, RWE can offer signals that can be tested more rigorously in an RCT.

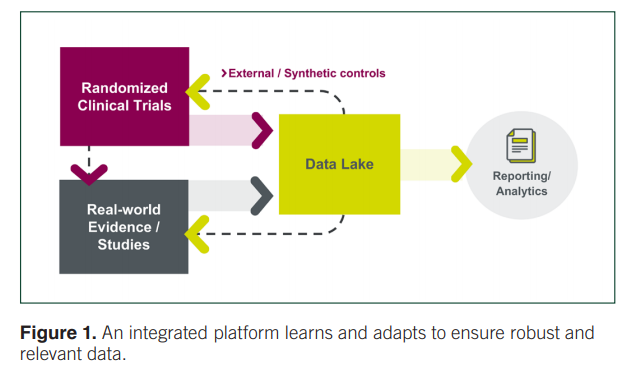
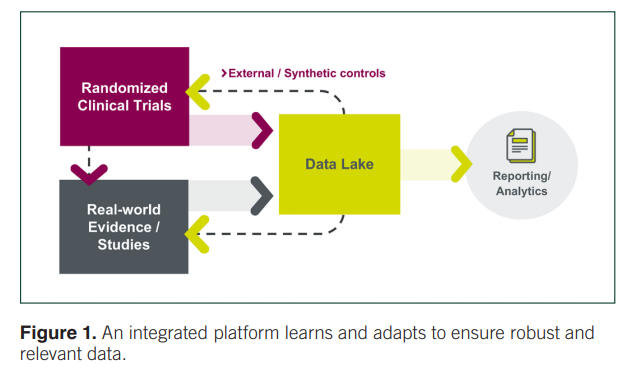
“Together, information from multiple data sources and studies all aggregated into one data lake can enhance our understanding and inform the research processes and patient care,” stated Leanne. “If we can achieve that, we’ll have taken a giant step toward what Amy Abernethy commented on last year, both now and beyond the pandemic.”
Leanne also explained that synthetic control arms offer a valuable way to advance drug development in this manner. Such controls can simplify patient enrollment and participation, eliminating the need to randomize patients to a control arm. All patients can be given the drug, so they can be assured that they will always receive the active treatment rather than a placebo. Only half as many patients will need to be enrolled, and more patients will be encouraged to participate thus driving faster enrollment.
“In the end, these benefits can mean that studies can conclude faster, advance the product quicker through the approval process, and get important and innovative new therapies out to patients rapidly,” stated Leanne, who then led the panel in a discussion of these topics.
REAL-WORLD APPLICATION OF SYNTHETIC
CONTROL ARMS
Leanne Larson (Parexel): MorphoSys recently completed FDA review and approval of an important new product using an external control to a phase two trial. Nuwan, can you tell us about that experience?
Nuwan Kurukulasuriya (MorphoSys): The goal of the L-MIND study was to assess an innovative chemo-free immunotherapy combination in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), which is a virulent and aggressive disease when untreated.
Fortunately, we observed high overall response rates and complete remission early in our phase two study, so we needed to find a way to accelerate the timelines for approval. We decided to deploy an external control strategy that involved cross-linking disparate real-world databases across the globe to generate a synthetic control arm to contextualize what was observed in the phase two setting. Our efforts paid off. Approximately nine months ago, we received a nod from the FDA with an approval and a precedent-setting label. The drug is now on the market and is already making a real difference in the lives of patients.
Leanne: What role do you see RWE playing in drug development today and how will this differ from the traditional model?
Nuwan: The future is bright. While randomized control trials give us a snapshot in time, RWE offers the possibility of assessing the patient’s experience dynamically, in motion, in real-time. What is truly exciting is the advancement of wearable technologies, artificial intelligence, machine learning, predictive analytics coupled with the maturation and sophistication of real-world data sets. This combination of tools will usher in a whole new era for clinical trials, accelerating execution, collapsing study timelines and ultimately culminating in improved patient lives.
Clinical heterogeneity is another challenge that RWE can help address. In some oncology clinical trials, it’s been estimated that only 3% of cancer patients are represented due to stringent inclusion and exclusion criteria. There’s also significant under-representation of minority patients in clinical trials, agnostic of therapeutic area. RWE holds the promise of broadening our focus, addressing these issues of inequity, and ultimately, bridging the “efficacy-effectiveness” divide.
Leanne: Over the years, it has been exciting to see how approaches to accessing and using real-world evidence have progressed as well as how the standards and regulatory approaches have evolved to ensure that we have robust studies. I think synthetic or external controls are really an interesting application of that going forward. What role do you think they might play in clinical trials moving forward?
Nuwan: In aggressive diseases, where patients may face a precipitous southward trajectory, speed is of the essence. It is imperative that such patients are afforded the medicines of tomorrow, today. Traditional full-blown, randomized, placebo-controlled trials may not only be too slow, but may also be considered unethical. Additionally, in rare genetic diseases, patient numbers may be too small to even entertain traditional clinical trial approaches. So the ability to explore how we can harvest the full value of real-world evidence and to find new and improved ways of running clinical trials by approximating, even mimicking randomization, is not a “nice to have”; it’s a must-have. I think progressive organizations should approach this with a sense of urgency and purpose because patients are waiting.
REGULATORY PERSPECTIVE
Leanne: Regulatory decision-making and processes for using external arms, such as synthetic controls, are new to most product developers. Amy, what do you see as the regulators’ perspective on external controls and RWE in product evaluation and how is it evolving?
Amy McKee (Parexel): Regulatory agencies worldwide are already using real-world data to support decision-making, as exemplified by the MorphoSys study Nuwan described. But at this point, regulators are primarily making one-off decisions.
So, where are we seeing this kind of real-world data use? Again, it has most often been used in areas of unmet need such as rare diseases and oncology.
How do we expand this use to more applications, more decisions, and more disease areas in general? Many regulatory authorities are working on frameworks or have already published guidance on how to use real-world data for regulatory decision-making. For instance, China has published and is already using its guidance. And very recently, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) announced two new guidances on the utilization of real-world data.
Leanne: This is an exciting area, and we are all looking forward to FDA releasing its guidance in the future. For now, what can product developers do to optimize this process? What are some of the challenges that FDA and other agencies might see to using this data?
Amy: It’s clear that we need to continue working on the detailed methodology around the capture and analysis of real-world data, and all of the regulatory agencies encourage companies to come in and discuss their proposals for using real-world data.
There are many different data sources that can be used, and one approach does not fit all circumstances. We’ve seen examples such as hybrid control arms and external control arms made up from electronic health records (EHRs) or from previously run clinical trials. Another approach is prospectively following patients in the EHRs concurrently with the investigational arm. Since it’s not a one-size-fits-all solution, we must continue to work on the detailed methodology that supports these varying ECA designs.
Regulatory agencies want to hear about plans to use RWE before receiving the submission, so that the methodology for the external control arm can be reviewed. The process can be very collaborative. Adding onto what Nuwan said, introducing more diversity into the patient group and data will only be a plus as we move forward.
Leanne: Yes, we’re still uncovering some of those benefits. I also appreciate your comments about engaging early with the agency when using synthetic controls. This was a central part of the MorphoSys L-MIND study.
REIMBURSEMENT FOR PRODUCTS STUDIED WITH RWE
Leanne: We’ve talked a lot today about the drug approval process, but that’s not the end of the story. Someone still needs to pay for these products. Many companies and product developers are still uncertain about what to expect from reimbursement and access decisions when RWE is involved, as well as how to prepare for and even influence these decisions. Sangeeta, are you aware of any payers that are using externally controlled trials as a basis for their decisions?
Sangeeta Budhia (Parexel): The focus of the payer viewpoint is on demonstrating value. Payers want to understand how a drug is going to impact patients in their healthcare system. Payers are looking for real-world effectiveness and comparative benefits along with economic value. Addressing the heterogeneity issue will be important to them as well.
Another thing that complicates reimbursement is the differences between countries and markets. Some markets, which we call the cost-effectiveness markets, look at the ratio of cost to effectiveness. Other markets, such as Germany and France, focus on comparative clinical effectiveness. They look at how does the treatment work in the real world compared to existing treatments for the particular patient population and then use that as the clinical benchmark for pricing negotiations. Spain and Italy look at the comparative clinical effectiveness and then evaluate the cost of treating the whole patient population. We have seen external or synthetic controls used as part of a reimbursement application in Europe, Australia and Canada, with mixed successes.
The key question has always been, “Does the data reflect the real world?” Synthetic or external control arms help address this question. I agree with Amy’s advice; Companies should engage with regulators and health technology assessment (HTA) bodies early on and discuss their data collection plan. In this way, they can reassure the payer that the data they are collecting is focused on reducing the uncertainty for them as well as for regulators. Payers are very sensitive to uncertainty and higher levels of risk, which are part of controlling the budget.
Leanne: What do payers think about real-world evidence as a whole?
Sangeeta: When cost effectiveness is the main decision driver, payers rely on a health economic model defined by the natural history of the disease. For example, in oncology, you will have a responder and a non-responder. We use natural history data sets to build the model and help define how patients would naturally progress through their disease. We’ve also used RWE to determine what happens in clinical practice. Payers want to understand the natural clinical practice for a patient and see how your drug can impact the way patients are currently being treated.
AUDIENCE Q&A
Lisa Henderson (Pharmaceutical Executive): Is it possible to adopt a U.S. strategy for HTAs in Europe?
Sangeeta: The short answer is yes, but the challenge is to demonstrate that U.S. data is relevant to the market you are entering, especially as HTA agencies are being established in countries beyond Europe. If you have collected data using a particular comparator, it’s important to ensure that the comparator is relevant to your target market. There are statistical techniques available to enable comparisons with comparators not in the clinical trials such as network meta-analyses, indirect treatment comparisons, and so forth. So, there are some ways to map data collected from external trials and within your clinical trial to data collected for comparators. However, this introduces a level of uncertainty and hence risk for the payer. As any company looks to the European market, they must address HTA challenges. So, I would recommend seeking early advice from HTAs, the EUnetHTA and individual agencies that provide early scientific advice services during the development of your phase 3 trial design. This can be done alongside early advice sought from regulators as well.
Lisa: What’s the best way to address the limitations or gaps in medical health record data? What are the pros and cons of retrospective versus prospective collection?
Leanne: As we’ve discussed today, there are pros and cons to using existing data versus generating the data prospectively. The important issue is finding a data source that’s robust enough to answer your questions and then understanding the data sources.
For oncology, for example, a significant amount of data sits in the note of EMRs. So, one approach is to look at those charts in real-time and pull out critical information. Another approach might be using natural language processing (NLP) to extract data from the notes and the unstructured fields in general. Our first question is always around what data already exists that we might be able to use.
There’s never one data source that can answer all of your questions fully and completely—the existing data were collected for a particular reason and the odds of that reason specifically matching yours are slim. The challenge, then, is to identify the right data sources, looking for the appropriate fit-for-purpose data to answer your questions, and then aggregating and analyzing it in a way that can answer your questions most effectively.
It’s a space that is evolving overall, and the technology is evolving around it. We have a rich set of data sources available globally, but exploration is required to understand exactly what’s in them, and to bring them together in a way that can best answer our questions. And then, of course, doing so within the context of the varying data privacy regulations around the world.
On the other hand, there is always the option to collect data in a prospective, observational, non-interventional study where you have more control over how you are collecting the data. We are finding that the most valuable approach is probably a hybrid of the two where we use as much secondary data as possible and aggregate it with what we collect prospectively. This process often gives us the richest, most-complete data set.
Amy: Looking at retrospective versus prospective data, one concern for the regulators is what has changed in terms of your specific disease context since these data were collected, particularly if you’re looking at retrospectively collected data. With some inborn errors in metabolism and genetic disorders where there’s no established standard of care and ancillary care hasn’t improved outcomes, you could probably go back fairly far and use retrospective data.
In contrast, in some oncology areas, new therapies have been approved so rapidly that you can’t go back six months and find patients who received the current standard of care. You have to ensure when designing your external control with real-world data that you closely examine the specific disease context. That’s one of the things about which regulators are very concerned. Are you truly comparing to what the standard of care is in that country? To that point, globally, there may be different standards of care. And that’s another thing to keep in mind as you’re constructing your external control arm.
Nuwan: I would encourage industry colleagues to plan ahead and think early about the customer view in the development process. Think about clinical development holistically and bring in the medical affairs perspective early in development. So, we shape and design not just randomized clinical trials, but also real-world evidence mechanisms, based on what we hear directly from the patient and the provider and the payer and the policymaker, all of the stakeholder groups that we have reviewed today.
Lisa: What can regulators do to further encourage and accelerate the use of synthetic control arms for pivotal research?
Amy: I think the regulators are working hard at trying to determine how best to use real-world data. As a community of stakeholders, we need to provide them with the best models and methodologies. You can’t answer every single question with real-world data that you perhaps could answer with a randomized trial. We’re not trying to replace randomized controlled trials. What we’re trying to do is find the best and fastest way to get helpful new therapies to patients.
This past year, COVID-19 really pushed the envelope. All of the agencies are looking to answer questions, both in terms of efficacy and safety, in the real-world data situation with this emerging public health issue.
Nuwan: A vacant area of opportunity for all of us is to think about end points differently. When we think about deploying real-world evidence approaches, we think about retro-engineering clinical endpoints that were designed for randomized clinical trials. As we move forward to push the envelope even further, a multi-stakeholder approach that benefits from the depth and breadth of richness that real-world data provides will be best. There’s a lot more to do in the space of endpoint development for real-world data.
Lisa: For studies involving a synthetic control arm as a comparator, how is selection bias avoided or addressed?
Leanne: It was really interesting to watch the development of the L-MIND study. Essentially, the process has been around one-to-one matching of these patients. It involved a very in-depth process for matching patients in the control arm, the synthetic control to the patients in the phase two trial, with nine different covariates in that particular study. That’s a pretty specific and intensive matching process and avoids the bias issue.
So, we’re not just taking large pools of patients and comparing them. We’re matching one-to-one each patient in the trial against the patients in the control population. It required accessing a significant volume of data, looking for patients with a rare disease that could match all nine of those covariates. On some level, I think it’s a bit more robust than if we had just enrolled the patient population and were looking for averages across the population and comparing those.
Lisa: How do the strategies discussed today differ for medical devices?
Amy: The strategies are very similar and in some regulatory agencies, they’re a little bit further ahead. The Center for Devices and Radiological Health (CDRH) published a guidance on how to use real-world data for device development in 2017. China approved a device for glaucoma based on real-world data already as well. I think devices are well on their way. Sometimes, they don’t get quite as much publicity as therapeutics, but they’re important, and the real-world guidance and approvals for devices that already exist support this.
For more information on RWE and External Control Arms:
- Watch the full panel discussion on-demand at https://www.pharmexec.com/view/can-synthetic-control-arms-address-regulator-and-hta-evidence-requirements-
- Download our Oncology eBook for further expert perspectives at https://explore.parexel.com/l/563702/2021-05-11/h8xk1d
1In a synthetic control arm, placebo arms are modeled based on pre-existing data instead of enrolling patients for a placebo group. Such data can include historical information, real-world data, a companion data set from another source.



