Cancer: From Rut to Racetrack
The objective is to make most cancers a curable, chronic condition. But with soaring costs, crowded therapeutic competition, and new diagnostic reimbursement challenges ahead, can industry deliver?
Cancer is science's moving target. Variations in the individual genotype have demonstrated that one size does not fit all and each patient's struggle is unique. Keeping pace with the relentless capacity of cancer to replicate its own destructive blueprint is one of the most formidable scientific hurdles of our time, with implications for everything from public health, safety, and prevention to the medical innovations that lead to treatments for many other life-threatening conditions.



(GETTY IMAGES / FUSE)
So the time is now: Where can we point to as the real areas of progress in the fight against cancer?
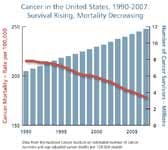
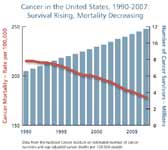
Cancer's high political profile and the commitment of the scientific community have produced real gains against cancer incidence and related mortality. US cancer death rates among men have decreased by more than one-fifth since 1990, largely due to advances in the treatment of lung, prostate, and colorectal cancers. Mortality among females has also dropped, but less rapidly, with the major gains concentrated in cancers of the breast and colon.
The key issue confronting researchers is that the overall record in fighting cancer remains mixed. Less progress has been made in saving or extending lives for cancers of the ovaries, liver, pancreas, and esophagus, whose incidence is increasing. Most clinical trials for new treatments still tend to demonstrate only marginal (two to three months) improvements in disease-free survival and/or overall survival rates. These two are the benchmark indicators of successful science that are attracting more scrutiny from the FDA in registering cancer medicines for clinical use.
Bombs Away with Chemistry
A continuing trend in the treatment of cancer is disabling tumor growth through toxic, undifferentiated interventions such as chemotherapy (antineoplastics).
Cancer therapies of the 20th century were largely a product of serendipity. In the 1940s during World War II came the discovery of alkylating agents (agents that damage DNA). Unrelated research found that US naval personnel exposed to nitrogen mustard gas evidenced major bone marrow suppression upon autopsy. It was hypothesized that this agent "targeted" certain types of cells whose nature it was to rapidly divide, and that this could potentially be useful to suppress malignant cells. Mechlorethamine thus became the first chemical agent to be approved by the FDA, in 1949.
Using a more rational method, Sidney Farber, known as the father of modern chemotherapy, studied the effects of folic acid on leukemic cell growth. Finding that folic acid stimulated these cancerous cells, he administered anti-folates to children with acute lymphoblastic leukemia and was able to induce remissions. His report on the findings in The New England Journal of Medicine was criticized; however, nearly a decade later, in 1956, administration of methotrexate led to the first cure of a metastatic cancer, choriocarcinoma.
Farber's work was important for other reasons. Scientific questions over his counterintuitive approach exemplify the intensity of debate among cancer specialists that continues to this day. In a field where access to a hypothesized "right" treatment can mean the difference between life and death, ethical concerns are equally as important as scientific judgment. Risk is a calculation that will always be factored in the basic bottom line of drug discovery—how long, and at what cost, can we commercialize a treatment so that its benefits reach patients? Balancing this equation is hardest in those with cancer.
As more chemotherapy agents came to market, combination chemotherapy was investigated and created a major breakthrough in cancer therapy. Administering multiple drugs with varying mechanisms of action makes it more difficult for the tumor to develop resistance. In addition, combining agents with individual efficacy and differing toxicity profiles may produce a synergistic effect and allow greater tolerability of the drug.
This in turn prompted changes in the approach used by investigators for drug therapy in the conduct of clinical trials and subsequently in clinical practice. For example, to prove single-agent activity, a logical trial would compare Drug A versus Placebo. If significant improvement is seen, then Drug A could be compared against the combination of Drug A + B. The following step is typically Drug A + B versus A + C. As demonstrated, the process became more complex, often with more than one study required.
Biology is Destiny
Chemotherapeutic agents prior to this point have all worked by inhibiting the metabolic pathways critical to cell division, and none were specific to the cancer cells. Because of the inherent ability of cancer cells to mutate and build resistance to this scattershot approach, a plateau has been reached in regard to clinical benefit.

Coming to the rescue is the increased understanding among researchers of cell biology, leading to the development of new lines of attack against cancer, particularly tumors that carry multiple mutations and show resistance in individuals very quickly. The emphasis today is on blocking mutated genes that fuel tumor growth, or on reprograming the DNA malfunctions that transform a normal cell into a cancerous one. In that sense, biology has succeeded chemistry as the necessary prerequisite for further progress against the disease.
The Right Drug for the Right Patient
Targeted therapies offer the potential to alter the molecular abnormality associated with the disease pathogenesis while reducing the effect on normal cells. The first targeted therapy was tamoxifen for treatment of breast cancer. In the 1960s, the estrogen receptor was described in breast tumors. Estrogen-blocking agents were studied in animal models in the early 1970s and tamoxifen, a selective estrogen-receptor modulator (SERM), gained FDA approval in 1977. It was later found to be most effective in patients whose tumors were considered to be estrogen-receptor or progesterone-receptor positive (ER/PR+). Tamoxifen has been shown to reduce the risk of recurrence or contra-lateral disease as well as to decrease the annual death rate. It continues to be widely used today.
The emphasis on targeted therapy has practical value beyond its scientific merits. Paired with a diagnostic assay, a targeted drug can demonstrate efficacy in those patients most likely to benefit. This can result in more efficient use of resources than the traditional shotgun approach of chemotherapy. Pairing the therapy with a diagnostic and then providing evidence that the intervention works in the specific patient population is often a difficult and costly exercise which adds to the risk of commercializing a promising compound.
Striking the Abnormal Chromosome
Therapies targeting constitutively active tyrosine kinases, or the specific chromosomal abnormality associated with a disease, have become a recent focus of research. Tyrosine kinase activation results in downstream cell signaling and cell proliferation. Various tyrosine kinases resulting from chromosomal abnormalities have been associated with the pathogenesis of many types of malignancies.
Chronic myelogenous leukemia (CML) was the first malignant disease found to be associated with a specific chromosomal abnormality. What is known as the "Philadelphia chromosome" encodes for the protein mutation bcr-abl and leads to uncontrolled tyrosine kinase activity. This chromosomal abnormality was first identified in 1961 when studying human leukemias and was then found to be consistent in patients with chronic myelogenous leukemia.
It was not until three decades later that a specific drug able to target this protein mutation was recognized. Imatinib mesylate (Gleevec) was identified from a library of small molecules which inhibit tyrosine kinases and found to decrease the number of bcr-abl cells formed in patients with CML. After further studies identified efficacy in patients who had failed standard treatment, the FDA granted accelerated approval of imatinib in patients whose CML had progressed after receiving interferon alfa. Not long after that, trials were conducted in which imatinib demonstrated improved efficacy over standard treatment as front-line therapy. As a result, imatinib has completely altered the course of CML, with long-term followup confirming an eight-year overall survival rate of 85 percent, with relatively few adverse reactions.
Encouraged by the success of imatinib, other agents targeting specific tyrosine kinases are being actively investigated for clinical use. The anaplastic lymphoma kinase (ALK) protein was first identified in anaplastic large cell lymphoma (ALCL) patients and is now the subject of intensive efforts in other malignancies.
In 2007, a fusion of the ALK tyrosine kinase with the echinoderm microtubule protein (EML4) was identified in a subset of non-small-cell lung cancer (NSCLC) patients. EML4-ALK occurs in approximately 2 percent to 7 percent of NSCLCs, most commonly in younger patients who have never smoked. Unlike the inhibition of bcr-abl tyrosine kinases, which took decades to achieve, crizotinib (sponsored by Pfizer; see "Planning Beyond the Petri Dish") was put into clinical studies within a few years of the identification of EML4-ALK. A Phase I study of crizotinib in patients who had already failed at least one prior therapy drug was recently published. Compared to traditional second-line therapy, which offers an average response rate of 10 percent, patients receiving crizotinib had an overall response of 57 percent, with 33 percent of patients having stable disease at a mean duration of 6.4 months. Crizotinib is now entering Phase III trials directly after Phase I comparing it to standard therapy in patients who have previously been treated with a platinum agent and also as first-line therapy. These studies only enroll patients with the specified mutation.
Despite these results, two secondary mutations conferring resistance to crizotinib have been described. This illustrates the challenge of commercializing a target therapy when tumor resilience may lead to declining efficacy around the initial breakthrough. From a public health perspective, the availability of follow-on therapies is critical to success in cancer treatment because of cancer's capacity to use the body's own defenses to induce resistance to a drug.
Monoclonal Antibody Therapy
The rise of monoclonal antibody [MaB] drugs is testament to the dominant role that molecular biology now plays as the pathway to new cancer treatments. These drugs work in various ways to either block the proliferation of growth receptors in cancerous cells or to impede the blood supply that nourishes tumors. Monoclonal antibody drugs now account for a sizable share of the oncology drugs market, with premium price points and sales that in a few cases exceed several billion dollars annually.
Bevacizumab (Avastin) is a humanized anti-VEGF (vascular endothelial growth factor A) monoclonal antibody. VEGF receptors play a critical role in the development of angiogenesis—the formation of new blood vessels—by stimulating the recruitment and proliferation of endothelial cells. By selectively binding VEGF—that is, inhibiting the binding of VEGF to its cell surface receptor—bevacizumab decreases microvascular growth of tumor blood vessels and limits the blood supply to tumor tissues. Bevacizumab currently has FDA-approved indications for use in four tumor types: colon, lung, kidney, and brain. Bevacizumab has also been studied in metastatic breast cancer and found to significantly improve progression-free survival, while not affecting overall survival. While improvements in progression-free survival initially were sufficient to gain FDA-accelerated approval for breast cancer, this indication was withdrawn by the FDA late last year and is currently up for debate in an appeal filed by the drug's sponsor, Roche. Further studies suggest there may be a subgroup of patients with tumors in which bevacizumab has greater impact.
Targeting: How Strong a Link?
As in all areas of research, better information allows for contradictions. Some—often unpublished—studies on targeted agents show negative results. The challenge now is to determine which subset of patients will benefit from these therapies. Alternatively, the goal is to determine who will not benefit, and therefore will be harmed by unnecessary toxicity and the avoidable costs of cancer therapy. Genotyping DNA from tumor blocks of breast cancer patients in the bevacizumab study mentioned previously found that specific VEGF-genotypes predicted a favorable median overall survival. Side effect issues are particularly important: the genetic variability of VEGF is also a predictor of clinically significant hypertension. Grade 3 and 4 hypertension was associated with improved median overall survival. Of course, these results still require validation, preferably in a prospective trial, as there are clear limitations to interpretation of retrospective evaluations of biomarkers.
Unlocking the Gene Code
With the completion of the Human Genome Project in 2003, new technologies to detect genomic alterations have become available. Scientists have begun to explore the complex genomes of tumors. Genome databases have been developed to assist in cataloging new discoveries. Next-generation sequencing has provided the ability to more efficiently compare tumor genomes to normal genomes of the same patient. Technology continues to work toward improving current techniques to provide increased information regarding tumor genomes at a decreased cost. Researchers are relying on genetic sequencing to determine which malignant pathways are active, regardless of where a tumor is located in the body.
Colorectal cancer (CRC) provided the first example of using this technology. The pathogenesis of metastatic colorectal cancer is characterized by sequential accumulation of both genetic and epigenetic (does not affect underlying DNA sequence) alterations. Ongoing research is focused toward translating the information regarding genomics research into clinically applicable predictive or prognostic tests.
Previous biomarkers that were developed to predict the efficacy of anti-epidermal growth factor receptor (EGFR) antibodies in patients with CRC exemplify the pitfalls of making assumptions about drug-target biology. Initial FDA approval of cetuximab for treatment of CRC included a requirement that there be positive immunohistochemical staining for the presence of EGFR in the tumor. It seemed reasonable that expression, and even perhaps the gene copy number, of the target would be required for efficacy, but this does not appear to be the case. EGFR expression itself does not predict response to EGFR monoclonal antibody therapy. EGFR-negative tumors have the potential to respond to cetuximab and thus EGFR immunohistochemistry before therapy is not warranted.
The Kras oncogene, which is detected in 40 percent to 45 percent of cases of CRC, is a key molecule in the effects of EGFR targeted therapy, as it is involved in an important downstream signaling cascade of EGFR. Activation of the Kras gene leads to a constitutive activity of the protein, resulting in continuous cell proliferation and other activities that promote carcinogenesis. When the Kras gene is mutated, degradation of the protein does not occur and activated Kras accumulates. This disturbed balance of Kras activation and degradation leads to resistance to EGFR blockage. The mutation status of Kras has been found to serve as a predictive factor for response to anti-EGFR-targeted therapy, such as cetuximab or panitumumab.
Thus, the research shows that administering EGFR-targeting monoclonal antibodies to unselected metastatic CRC patients can no longer be considered the standard of care, as those agents will be ineffective in patients with activating mutations in Kras. The test to determine mutational status is not yet confirmed. Despite the fact that mutations are binary events with less room for interpretation than protein or mRNA expression-based tests, a balance between accuracy and practicality is still needed. A number of options for mutation status testing are available, with all methods based on polymerase chain reaction (PCR) testing platforms. Concerns include the difficulty of obtaining tumor samples suitable for molecular analyses (for example, a concentration of mutant copies high enough to detect). In addition, the genetic heterogeneity of CRC means that the absence of detectable Kras mutations in the primary tumor cannot formally exclude the presence of a Kras mutation in metastases.
Nevertheless, panitumumab for the treatment of patients with uncontrolled, or "wild-type," Kras tumors is the first example of the approval of a drug therapy for solid tumors that is based on a genetic test. It opens a new era in biomarker-driven therapy in this disease.
Combination Targeted Therapy
The combination of cytotoxic chemotherapy allows further cancer destruction and decreases tumor resistance. Hence it could be a valid strategy in targeted molecular biologic therapies as well. But the results from trials appear inconclusive. In one study, the combined effect of the MaB drug cetuximab and the current standard of care in metastatic colorectal cancer, bevacizumab plus flouropyrimidine-based chemotherapy, resulted in a rate of progression-free survival that was significantly shorter in the combined antibody therapy arm. The conclusion was that combining multiple forms of targeted therapies, specifically anti-EGFR and anti-VEGF, may not be analogous to combining different types of chemotherapy, due to subtle interactions in intracellular signaling.
If combination monoclonal antibody therapy is not yet tagged as the next magic bullet, most drug companies are making it the cornerstone of their research programs in cancer, accounting for the majority of the more than 800 anti-cancer compounds that Pharmaceutical Research and Manufacturers of America (PhRMA) members have under development. Progress continues: A recent study by The Ohio State University is optimistic about the combination of a targeted PI3K inhibitor with an agent that reverses the epigenetic changes that cause gene silencing.
All this science is intuitive. Cancer development may involve either or both of these pathways—abnormal genes (or "oncogenes") that regulate cell growth become activated and/or the genes that prevent cancer development (or "tumor-suppressor genes") are silenced or turned off. More than 20 PI3K-targeted inhibitors have recently been introduced in clinical trials. More striking, though, is that perhaps success of these agents is only achieved when administered in combination with epigenetic drugs for synergistic effects. The recent study at Ohio State demonstrated activation of PI3K/AKT does play a role in epigenetic silencing and lays the foundation for future therapies targeting both of these mechanisms.
Another new innovation unleashed by mapping the genome is the role that damage to DNA plays in destroying healthy cells. Poly (ADP-ribose) polymerase, or PARP, is an enzyme which repairs damage done to our DNA by radiation or cell mutations. PARP-1 inhibitors are in a new class that causes inhibition of PARP function. BSI 201 and olaparib were the first of many now being studied. Initial clinical trials showed efficacy in breast, ovarian, and prostate cancer. BSI-201 has shown efficacy in triple-negative breast cancer, a type of cancer that unfortunately does not have many therapeutic options. These drugs represent a new investigational option to treat BRCA gene-mutated cancers.
PARP inhibitor drugs show promise for two reasons: they demonstrate significant anti-tumor effects in several types of cancer and have less side effects. Of note, these agents may also be effective in non-BRCA-mutated tumors. These agents are now in investigation for a wide range of malignancies, including uterine and brain tumors, lymphomas, and chronic leukemias.
Vaccine Fix
As research continues to uncover additional mechanisms to promote tumor cell death, vaccines are emerging as a new weapon—not only to prevent cancer but to combat it as well. The most well-known breakthrough in this area is the use of vaccines to prevent certain strains of the human papilloma virus (HPV), which have been associated with cervical cancer. Most recently, vaccines are being investigated for their potential to treat active cancer. As with all cancer vaccines, the aim is to generate an immune reaction against a specific molecular target that is greater than the tumor's immunosuppressive mechanisms. The first therapeutic cancer vaccine in the US to be approved was sipuleucel-T in 2010. This autologous vaccine demonstrated an increase in median overall survival from 21.7 months to 25.8 months in patients with metastatic castration-resistant prostate cancer. However, the extra 4.1 months of survival does not come without an extravagant preparation process and costs. Physicians also struggle with determining when continuation of therapy is worthwhile.
Immunotherapy and vaccine research continues to progress with ongoing Phase III trials in the areas of NSCLC and non-Hodgkin's lymphoma (NHL) as well as further explorations of combination therapy with vaccines as well as cytotoxic agents.
New Hurdles to Approval
An ethical debate that will impact the future climate for cancer research is whether new oncologic agents should undergo accelerated approval prior to adequate, well-controlled studies, or if these agents should only be available on a compassionate-use basis. The average time for a drug to gain regular FDA approval is seven years from the start of human trials, and approval is usually based on survival advantages for the new agent. Accelerated approval was created in 1992 as a method for patients to gain earlier access to promising drugs for diseases with limited treatment options. Accelerated approval can be obtained by demonstrating improvements in surrogate endpoints, such as tumor progression, progression-free survival, response rates, or symptom relief in Phase II or Phase III trials.
Adequate, well-controlled trials must then be conducted, and if a survival benefit is demonstrated, the agent can be converted to regular FDA approval. The problem is that it is increasingly apparent that accelerated approval is no longer a carte blanche—regulators are getting tougher in demanding that efficacy and safety be certified through real-time clinical use to obtain permanent licensing rights. More restrictive labeling is another consequence of the emphasis on survival benefits. All told, the trend adds to the risk inherent in the registration process, which means that the market access hurdle for cancer drugs will rise higher. At present, an anti-cancer drug entering Phase I testing has only a 3 percent chance of entering the market, compared to the 10 percent average for drugs overall.
Between 1992 and 2010, 35 oncology drugs with 47 new indications were granted accelerated approval. Of these, 28 indications have been converted to regular approval. The first FDA-proposed withdrawal of an accelerated approval indication was made in late 2010 for bevacizumab in metastatic breast cancer. This decision was made after required confirmatory trials failed to substantiate the improvements in progression-free survival seen with earlier studies. Roche, as sponsor, is expected to undergo a hearing with the FDA in June to determine the future of this indication.
Twenty-one accelerated approval oncology indications have not been converted to regular approval, as confirmatory trials have not yet been completed. The FDA Amendments Act of 2007 allows the FDA to fine companies up to $10 million for not completing confirmatory trials in a timely manner.
One challenge in confirmatory trials after accelerated approval is patient accrual. Patients are reluctant to risk randomization to standard therapy after hearing reports of promising therapeutic alternatives and having off-study access to such agents. To overcome this obstacle, the FDA has recommended sponsors submit interim Phase III trial data for accelerated approval with continued followup of current patients for confirmatory studies. The FDA is also encouraging open discussion of the timeline and plans for confirmatory studies.
Themes for the Future
Pharmacogenomics, the study of the role of inherited and acquired genetic variation in drug response, is in the forefront of personalized medicine for cancer. It facilitates the identification of biomarkers that can help optimize drug selection, dosage, treatment duration, and avert adverse drug reactions.
However, just where pharmacogenomics fits into clinical practice is still evolving. Although the technology holds promise for the future, the ability to fulfill that potential will depend on the characterization of tissue and blood samples, collected from well-differentiated and uniformly treated patient populations, using validated and standardized assays. As new technology requires smaller amounts of tissue for diagnosis, it will be a challenge to maintain tissue banks, which can be utilized for future areas of investigation. And the clinical value of these gene sets must ultimately be validated in prospective clinical trials. In contrast to earlier data where the currently studied biomarkers are being validated retrospectively with bevacizumab in breast cancer (VEGF) and cetuximab in colon cancer (Kras), enrollment in studies for current promising breakthroughs such as crizotinib requires upfront biomarker tests for mutations (EML4-ALK).
The landscape of cancer treatment is changing rapidly as more underlying molecular abnormalities, which play a role in prognosis as well as response to various therapies, become apparent. While anatomical staging of tumors will always be a treatment mainstay, further classification based on the genetic makeup of tumors will hopefully carry more prognostic weight and influence clinical decisions. It is likely we will see more FDA-mandated pharmacogenomic information in package inserts as we further identify genetic variations that affect medications' efficacy and toxicity. Although payers may drag their heels initially, reimbursement for pharmacogenomic testing will be a requirement that precedes reimbursement for these medications. Through all of these changes, the utility of a pharmacist will be essential, as drug dosing will not only be based on body measurements but on molecular profiles as well.
This wealth of growing knowledge will further enable development of personalized cancer therapy based on a tumor's molecular profile. Outcomes can be improved and side effects avoided by utilizing individual genetic information to determine the optimal anti-neoplastic therapy for patients. This genetic information can further aid the development of novel targeted therapies and should enhance our search for understanding mechanisms of resistance and predictive biomarkers. Cancer is where the future of personalized medicine will play out. It will be a strong focus of research and has the potential to pilot new breakthroughs.
Natalie Dickmeyer, PharmD, is a researcher at the Indiana University Simon Cancer Center. She can be reached at ndickmey@gmail.com.
Lindsay Rosenbeck, PharmD, is a researcher at Indiana University Health. She can be reached at lrosenb1@iuhealth.org.




















The Misinformation Maze: Navigating Public Health in the Digital Age
March 11th 2025Jennifer Butler, chief commercial officer of Pleio, discusses misinformation's threat to public health, where patients are turning for trustworthy health information, the industry's pivot to peer-to-patient strategies to educate patients, and more.


Navigating Distrust: Pharma in the Age of Social Media
February 18th 2025Ian Baer, Founder and CEO of Sooth, discusses how the growing distrust in social media will impact industry marketing strategies and the relationships between pharmaceutical companies and the patients they aim to serve. He also explains dark social, how to combat misinformation, closing the trust gap, and more.



